Brief communication (behind paywall) Yleaf: software for human Y-chromosomal haplogroup inference from next generation sequencing data, by Arwin Ralf, Diego Montiel González, Kaiyin Zhong, and Manfred Kayser, Mol Biol Evol (2018), msy032.
Abstract
Next generation sequencing (NGS) technologies offer immense possibilities given the large genomic data they simultaneously deliver. The human Y chromosome serves as good example how NGS benefits various applications in evolution, anthropology, genealogy and forensics. Prior to NGS, the Y-chromosome phylogenetic tree consisted of a few hundred branches, based on NGS data it now contains many thousands. The complexity of both, Y tree and NGS data provide challenges for haplogroup assignment. For effective analysis and interpretation of Y-chromosome NGS data, we present Yleaf, a publically available, automated, user-friendly software for high-resolution Y-chromosome haplogroup inference independently of library and sequencing methods.
Here is a link to the software Yleaf’s website, from the Department of Genetic Identification, at the University of Erasmus Medical Center.
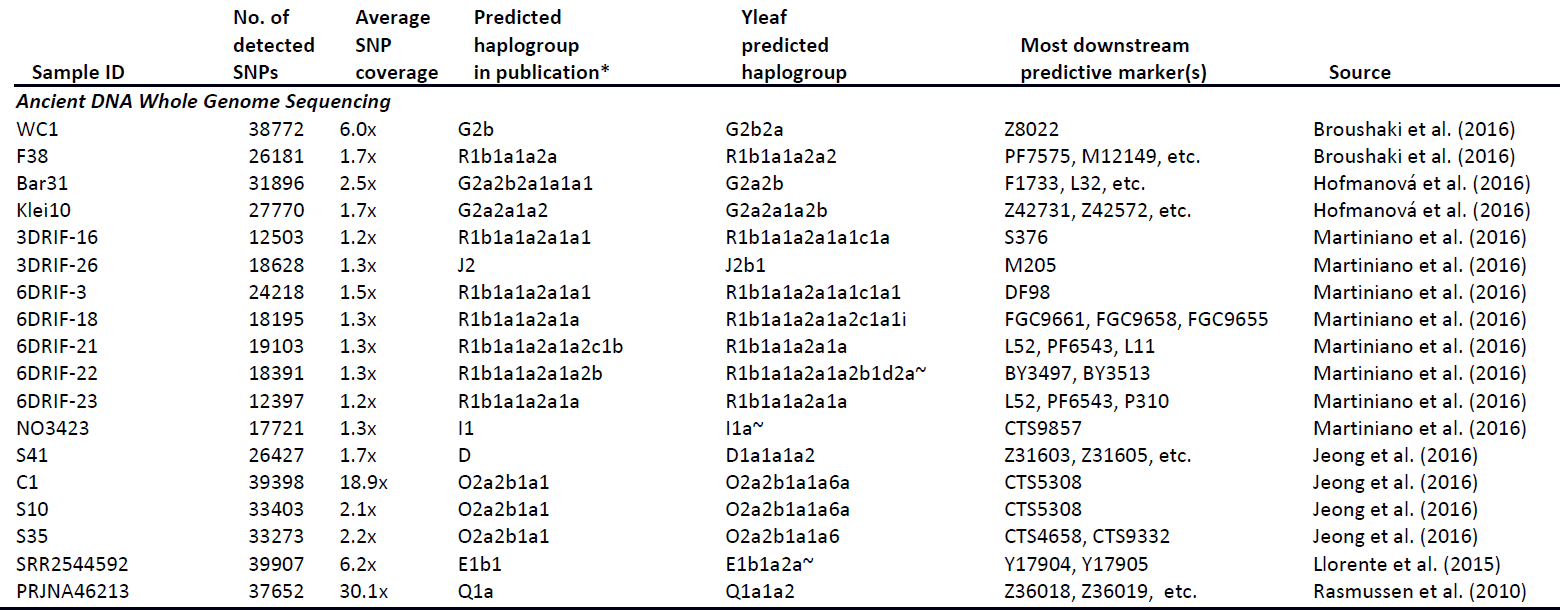
Excerpt:
In the time of NGS (or massively parallel sequencing, MPS), the amount of genomic data produced and made publically available is rapidly expanding, providing valuable resources for many areas of research and applications. Due to its haploid nature and male-specific inheritance, the non-recombining part of the human Y-chromosome (NRY) is highly suitable for phylogenetic studies and for addressing questions in evolution, anthropology, population history, genealogy and forensics (Jobling & Tyler-Smith, 2017). Over recent years, NGS data allowed the phylogenetic NRY tree to dramatically increase in size and complexity (Hallast et al. 2014; Poznik et al. 2016). The two most comprehensive tree versions ISOGG (http://www.isogg.org/tree) and Yfull (https://www.yfull.com/tree) currently contain thousands of branches. However, the complexity of both, Y tree and NGS data provide immense challenges for NRY haplogroup assignment, which reflects a key element in many NRY applications. Here we introduce Yleaf, a Phyton-based, easy-to-use, publically-available software tool for effective NRY single nucleotide polymorphism (SNP) calling and subsequent NRY haplogroup inference from NGS data. By comparative whole genome data analysis, we demonstrate high concordance of Yleaf in NRY-SNP calling compared to well-established tools such as SAMtools/BCFtools (Li et al. 2009), and GATK (McKenna, et al. 2010) as well as improved performance of Yleaf in NRY haplogroup assignment relative to previously developed tools such as clean_tree (Ralf et al. 2015), AMY-tree (Van Geystelen et al. 2015), and yHaplo (Poznik, 2016).
Yleaf allows analyzing NRY sequence data from many types of NGS libraries i.e., whole genomes, whole exomes, large genomic regions, and large numbers of targeted amplicons. Several modifications relative to our previously developed clean_tree tool (Ralf et al. 2015) were implemented to optimize the performance especially relevant for extremely large NGS datasets such as whole genomes. For instance, Yleaf extracts the Y-chromosomal reads prior to further processing and uses multi-threading, a batch option is included too. Importantly, Yleaf provides drastically increased haplogroup resolution i.e., from Downloaded from 530 positions defining 432 NRY haplogroups with clean_tree (Ralf et al. 2015) to over 41,000 positions defining 5353 haplogroups with Yleaf. For a detailed method description see the supplementary material.
Featured image: From Martiniano et al. (2017).
Related:
- Population substructure in Iberia, highest in the north-west territory (to appear in Nature)
- Analysis of R1b-DF27 haplogroups in modern populations adds new information that contrasts with ‘steppe admixture’ results
- Migrations painted by Irish and Scottish genetic clusters, and their relationship with British and European ones
- Y-DNA relevant in the postgenomic era, mtDNA study of Iron Age Italic population, and reconstructing the genetic history of Italians
- The Tollense Valley battlefield: the North European ‘Trojan war’ that hints to western Balto-Slavic origins
- Olalde et al. and Mathieson et al. (Nature 2018): R1b-L23 dominates Bell Beaker and Yamna, R1a-M417 resurges in East-Central Europe during the Bronze Age